
Translate this page into:
The usual suspects: A case series on bilateral astrocytic hamartomas in tuberous sclerosis

*Corresponding author: Hitisha Mittal, Department of Ophthalmology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, India. hitishamittal72@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Satish NM, Guliani NC, Rajshekhar V, Mittal H, Mehta A. The usual suspects: A case series on bilateral astrocytic hamartomas in tuberous sclerosis. J Ophthalmic Res Pract. doi: 10.25259/JORP_36_2024
Abstract
Hamartomas are non-malignant growths composed of abnormal cells in normal locations. Various hamartomas are seen in tuberous sclerosis (TS), a rare multisystem genetic disorder commonly involving the skin, brain, lungs, heart, kidneys, and eyes. It results from TS complex (TSC1) or TSC2 gene mutation that may occur sporadically or be inherited in an autosomal dominant pattern. We present three cases of TS with retinal hamartomas. These hamartomas usually remain undetected until they either cause breathing difficulties or TS-associated neuropsychiatric disorders. Angiomyolipomas and rhabdomyomas can also occur in TS and should be thoroughly investigated, as they pose a risk of fatal rupture and hemorrhage. The purpose of this case series is to familiarize an ophthalmologist with various presentations of TS and stress on importance of careful clinical examination of TS patients.
Keywords
Angiofibroma
Astrocytoma
Neurocutaneous disorder
Retinal hamartoma
Tuberous sclerosis

INTRODUCTION
Tuberous sclerosis (TS) is a rare, genetic neurocutaneous disorder with hamartomas in multiple organs. It may be sporadic or autosomal dominant due to mutation in either the TS complex (TSC1) gene encoding Hamartin on chromosome 9q34 or TSC2 encoding Tuberin on chromosome 16p13.[1] Vogt initially described it as a triad of mental retardation, seizures, and facial angiofibroma. However, this triad was found in only 30% of patients with most having normal intelligence.[2] The diagnosis of TS is based on clinical criteria [Table 1][3] but a mutation detected in TSC1 or TSC2 is sufficient for diagnosis regardless of clinical features.[4]
|
|
| Definite diagnosis: Two major features or one major feature with ≥2 minor features. Possible diagnosis: Either one major feature or ≥2 minor features. | |
DNA: Deoxyribonucleic acid
$ Note that 10% to 25% of TSC patients have no mutation identified by conventional genetic testing, and a normal result does not exclude TSC, or have any effect on the use of clinical diagnostic criteria to diagnose TSC.
CASE SERIES
Case 1
A 15-year-old female with genetically confirmed TS was referred to ophthalmology for routine examination. Her best-corrected visual acuity (BCVA) was log 0 in both eyes. She had facial angiofibroma, ash leaf macules, and molluscum pendulum [Figure 1a]. She also had a subungual fibroma [Figure 1b]. On ocular examination, the anterior segment was within normal limits. Fundus examination revealed a white translucent retinal lesion with indistinct margins at the posterior pole of both eyes [Figure 1c]. One of the lesions had a sclerosed vessel over the surface. On spectral domain (SD) optical coherence tomography (OCT), multiple dome-shaped hyperreflective lesions were seen in the retinal nerve fiber layer (RNFL) [Figure 1d and e]. On OCT angiography, an intricate network of vasculature was noted [Figure 1e]. Systemic imaging revealed multiple cortical tubers and subependymal nodules in the brain. Contrast-enhanced computed tomography (CECT) of the chest and abdomen revealed multiple nodular lesions in the lungs and possible angiomyolipoma in the liver and both kidneys [Figure 1f].
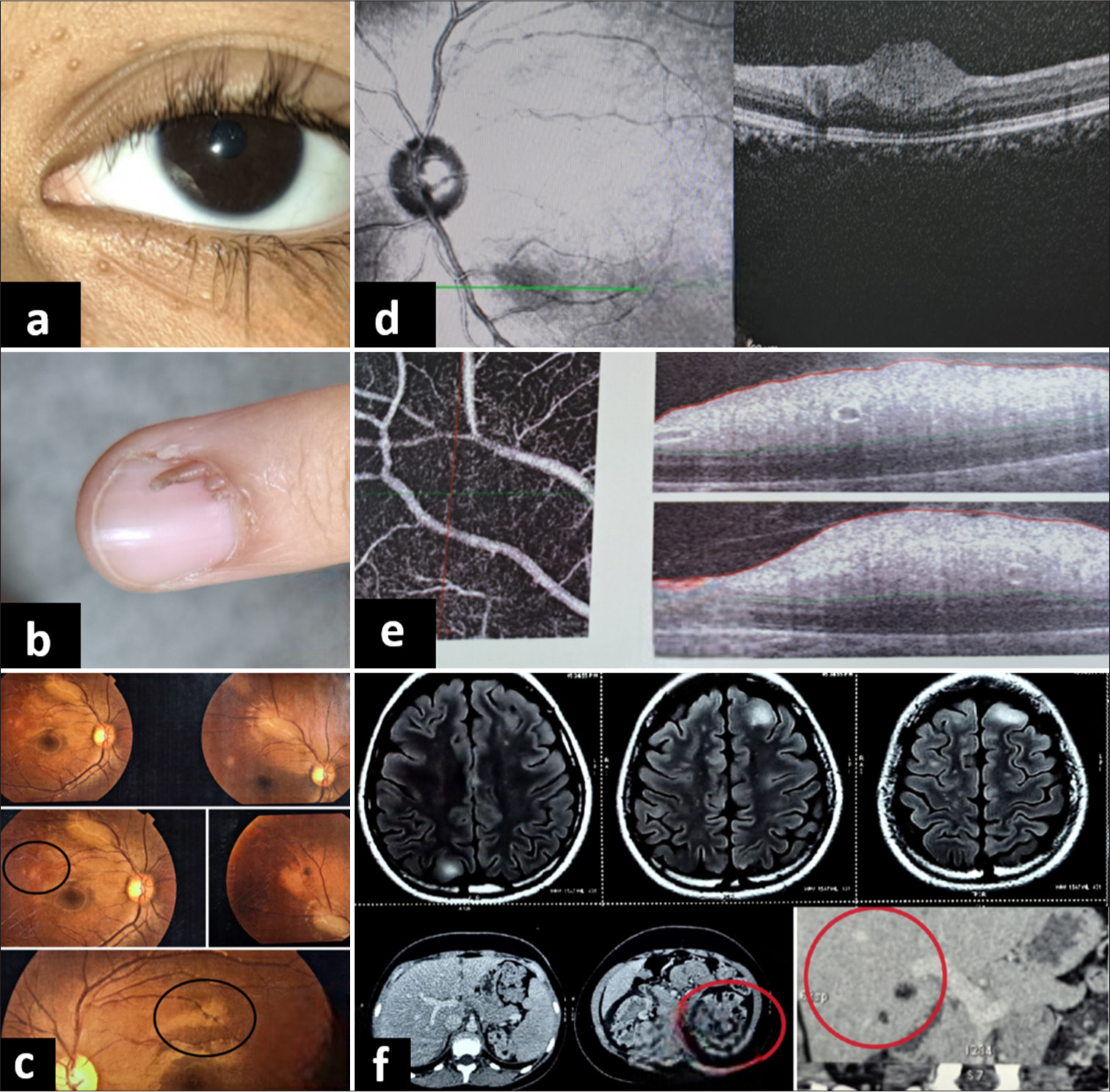
- (a) Molluscum pendulum lesions on eyelids, (b) sub ungual fibroma, (c) white translucent retinal lesions with indistinct margins at the posterior pole of both eyes, (d) and (e) dome shaped hyperreflective lesions in retinal nerve fibre layer on spectral domain optical coherence tomography, and (f) contrast-enhanced computed tomography chest and abdomen showing multiple nodular lesions in lungs suggestive of lymphangiomatosis (red circles) and possible angiomyolipoma in liver and both kidneys.
Case 2
An 8-year-old female was referred from school for regular examination. On general physical examination, angiofibroma, shagreen patches, and ash leaf macules were noted [Figure 2a]. With a BCVA of log 0 in both eyes, the anterior segment examination was normal. Fundus examination showed two different types of lesions: A flat salmon-colored translucent lesion with indistinct margins in the right eye and a larger whitish translucent lesion in the left eye [Figure 2b]. OCT revealed a similar appearance of lesions [Figure 2c], as seen in Case 1. Magnetic resonance imaging (MRI) of the brain revealed subependymal nodules and cortical tubers [Figure 2d].
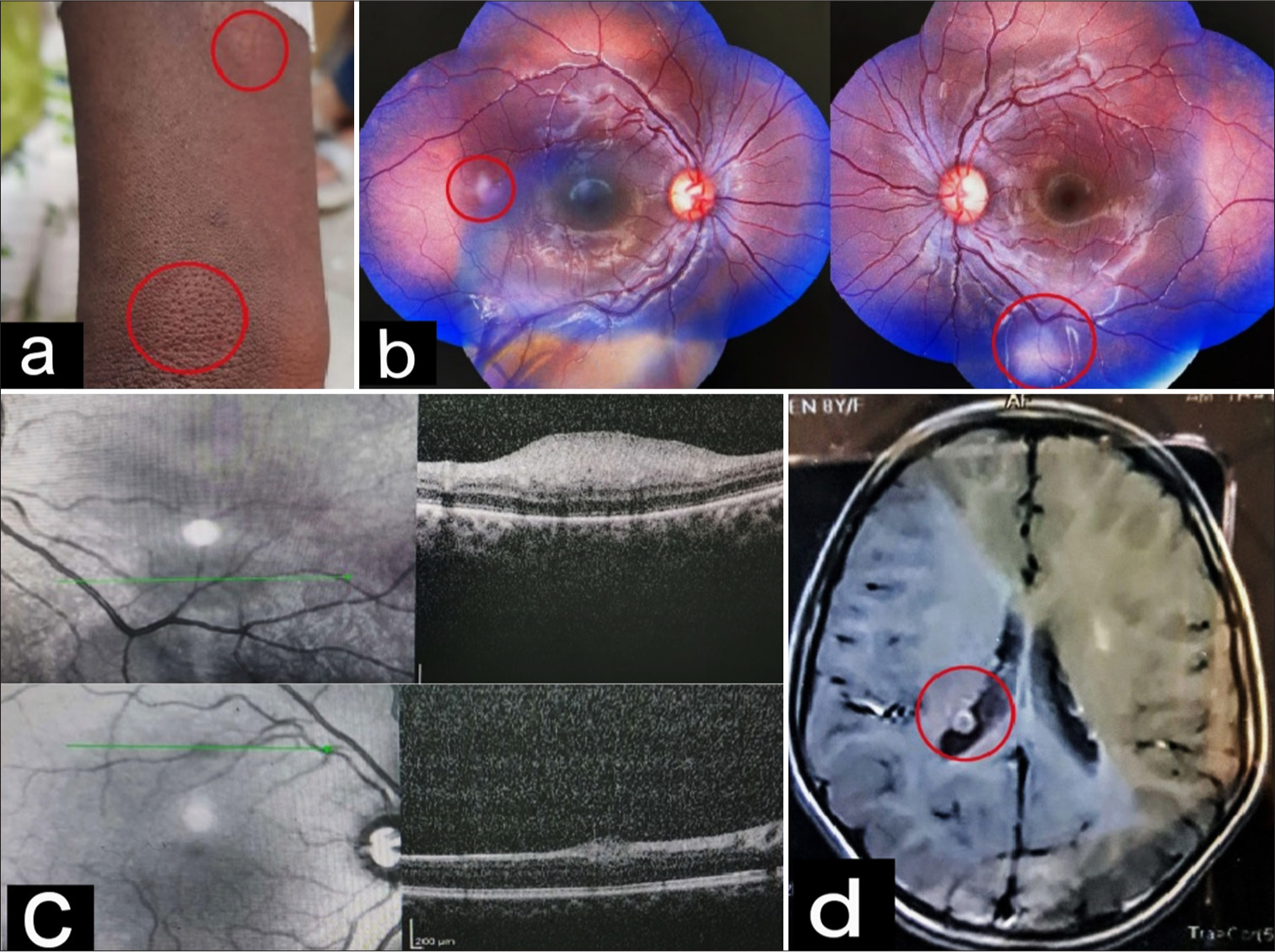
- (a) Shagreen patches and ash leaf macules on the forearm (as marked in red circle), (b) a flat salmon-colored translucent lesion with indistinct margins in the right eye and a larger whitish translucent lesion in the left eye (as marked in red circles), (c) dome-shaped hyper-reflective lesions in retinal nerve fiber layer on spectral domain-optical coherence tomography, and (d) magnetic resonance imaging brain showing subependymal nodules and cortical tubers (as marked by red circle).
Case 3
A 10-year-old boy presented with complaints of itching in his eyes. On examination, he had multiple facial angiofibromas [Figure 3a] and hypopigmented ash leaf macules [Figure 3b]. On ocular examination, his BCVA was log 0 (with +2 diopter sphere acceptance), and he had perilimbal hyperpigmentation in both eyes. Fundus examination revealed small white lesions in the retinal periphery of both eyes, resembling the hamartomas seen in other patients [Figure 3c]. On OCT, similar dome-shaped hyperreflective lesions were seen in RNFL [Figure 3d]. MRI brain showed two small cortical tubers [Figure 3e].
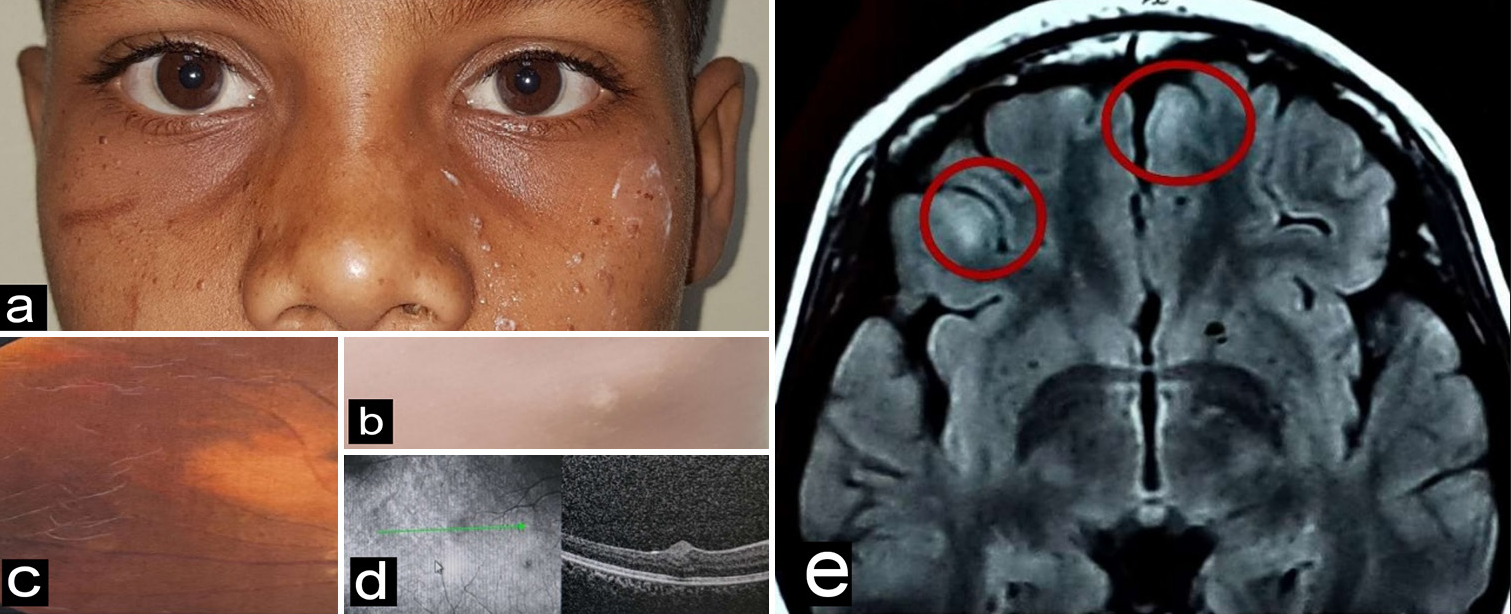
- (a) Multiple facial angiofibromas, (b) hypopigmented ash leaf macule, (c) a flat white translucent lesion with indistinct margins, (d) dome-shaped hyperreflective lesion in retinal nerve fiber layer on spectral domain-optical coherence tomography, and (e) Magnetic resonance imaging brain showing two small cortical tubers (as marked by red circles).
DISCUSSION
Retinal hamartomas are the most common ocular manifestations of TS. Three different patterns of retinal hamartomas have been described. The most common type (seen in 50% of TS patients) is a flat yellow translucent lesion with indistinct margins often found near the ends of the arcades.[5] The second type is a white opaque multinodular lesion resembling mulberry, and the third type is a combination of the above two, known as the transitional type. These are less common.[5] Retinal hamartomas can be bilateral in 30% of patients. In this series, all patients had a bilateral presentation with flat translucent retinal hamartomas, whereas two of them also had the third transitional type.
On SD-OCT, the flat type of hamartoma is seen as a dome-shaped elevation with hyper-reflectivity in RNFL, whereas the type two hamartomas usually show elevated hyperreflective masses with disorganization and moth-eaten spaces with posterior shadowing.[5]
Tubers found in various organs in TS have high angiogenic properties and vascular endothelial growth factor expression. Shields et al. reported four patients with aggressive retinal hamartomas associated with serous retinal detachment and retinal hemorrhages.[6] Thus, it was important to explain to each patient the possible progression of these seemingly benign lesions. Treatment options include laser photocoagulation of hamartomas, intravitreal bevacizumab with or without triamcinolone for macular edema, and enucleation for a painful blind eye.[7,8]
Other ocular features of TS include colobomas of the iris and choroid, iris hamartoma, angiofibroma of eyelids, and ciliary epithelium hamartoma.[4] Papilledema can be seen in obstructive hydrocephalus secondary to subependymal glial cell astrocytoma (SEGA). Optic nerve head hamartomas, nerve palsies, cortical visual impairment, and visual field defects are other manifestations.[9] Those with TSC2 mutations are more likely than TSC1 to have retinal astrocytic hamartomas, angiomyolipomas, renal cysts, infantile spasms, SEGA, and intellectual disability, hence the importance of genetic counseling in such patients.
Neurological lesions in TS include cortical tubers, subependymal nodules, or giant cell astrocytoma.[9] All three of our patients had cortical tubers, whereas two of them had subependymal nodules. Cortical involvement is common in patients with retinal hamartomas.
Dermatological features may be the first noticeable lesions in TS. Ash leaf macules were seen in all of our patients, shagreen patches in two, and subungual fibroma in one patient.
Renal angiomyolipoma or renal cysts are found in 70– 80% of TS patients. It was also seen in one of our patients. Interestingly, renal involvement is more common in older TS patients than in children.[4,5] Pulmonary lesions include lymphangiomatosis, which may be asymptomatic or cause dyspnea.[2,4] One of our patients had this asymptomatic finding on the CECT chest.
Sirolimus and Everolimus are approved for SEGA, angiomyolipoma, and intractable epilepsy treatment.[10] All three of our patients were asymptomatic but need regular ophthalmic follow-up due to the risk of serous retinal detachment. Furthermore, the presence of neurological, renal, and pulmonary lesions warrants lifelong follow-up to prevent any long-term sequelae.
CONCLUSION
Hamartomas in TS can affect multiple organs. The incidental detection of retinal astrocytic hamartomas by an ophthalmologist should prompt a detailed systemic investigation for other manifestations of TS, which will help in the timely detection of life-threatening manifestations and improve the patient’s quality of life. Further, these patients need genetic counseling and regular follow-up.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Genetics of tuberous sclerosis complex: Implications for clinical practice. Appl Clin Genet. 2016;10:1-8.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiovascular manifestations of tuberous sclerosis complex and summary of the revised diagnostic criteria and surveillance and management recommendations from the International Tuberous Sclerosis Consensus Group. J Am Heart Assoc. 2014;3:e001493.
- [CrossRef] [PubMed] [Google Scholar]
- Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-54.
- [CrossRef] [PubMed] [Google Scholar]
- Ophthalmic manifestations of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:17-25.
- [CrossRef] [PubMed] [Google Scholar]
- Review of spectral domain-enhanced depth imaging optical coherence tomography of tumors of the retina and retinal pigment epithelium in children and adults. Indian J Ophthalmol. 2015;63:128-32.
- [CrossRef] [PubMed] [Google Scholar]
- Aggressive retinal astrocytomas in four patients with tuberous sclerosis complex. Trans Am Ophthalmol Soc. 2004;102:139-47. discussion 147-8
- [Google Scholar]
- Photodynamic therapy with verteporfin to induce regression of aggressive retinal astrocytomas. Acta Ophthalmol. 2008;86:794-9.
- [CrossRef] [PubMed] [Google Scholar]
- Combined bevacizumab and triamcinolone acetonide injections for macular edema in a patient with astrocytic hamartomas and tuberous sclerosis. Ophthalmic Surg Lasers Imaging Retina. 2013;44:85-90.
- [CrossRef] [PubMed] [Google Scholar]
- Neuroophthalmological manifestations of tuberous sclerosis: current perspectives. Eye Brain. 2019;11:13-23.
- [CrossRef] [PubMed] [Google Scholar]
- Sirolimus for angiomyolipoma in tuberous sclerosis complex of lymphangioleiomyomatosis. N Engl J Med. 2008;358:140-51.
- [CrossRef] [PubMed] [Google Scholar]




