
Translate this page into:
Eye involvement in lipoid proteinosis: A rare clinically forgotten entity

*Corresponding author: Raisa Arora, Department of Ophthalmology, Employee’s State Institute and Post Graduate Institute of Medical Sciences and Research, Basaidarapur, Delhi, India. drraisatandon@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Arora R, Kohli V, Kumar V. Eye involvement in lipoid proteinosis: A rare clinically forgotten entity. J Ophthalmic Res Pract 2023;1:76-8. doi: 10.25259/JORP_31_2023
Abstract
Lipoid proteinosis (LP), also known as Urbach-Wiethe disease, is a rare autosomal recessive genodermatosis having multisystem syndromic involvement of skin, upper aerodigestive tract, internal organs, and eyelids. Although the ocular manifestations are rare, it is important to identify these clinical entities for early diagnosis and improved outcomes. We, hereby, describe a case of a 17-year-old female who presented with complaints of chronic itching and discomfort of both eyelids on and off.
Keywords
Lipoid proteinosis
Eyelids
Moniliform blepharosis

INTRODUCTION
Lipoid proteinosis (LP), also known as Urbach-Wiethe disease, is a rare autosomal recessive genetic disorder described first in 1929; fewer than 300 cases have been reported in the literature to date.[1]
It is a multisystemic disorder with the predominant involvement of skin and mucosal membranes of the respiratory and digestive tracts. It is characterized by early infancy onset and presents with a weak cry and hoarse voice due to infiltration and involvement of the larynx.[2]
LP is histologically characterized by deposits of periodic acid Schiff-positive hyaline-like material intracellularly in the substance of the skin, mucous membranes, and the involved organs. Genetic defects attributed to this disease are the mutations in the gene encoding extracellular matrix protein 1 on chromosome 1q21 leading to a loss in key protein-protein interactions.[3] This gene mutation is rare and <20 variants have been reported in the literature, making it an extremely rare entity.
Although ocular involvement in LP is found to be rare, ophthalmologists may encounter various ocular complications accompanying this syndrome in clinical practice.
CASE REPORT
A 17-year-old young female presented to our outpatient department with complaints of itching and discomfort of eyelids in both eyes on and off for 9 years. There was no history of redness, watering, or discharge. There is no family history of a similar disease. On detailed ocular examination, her unaided visual acuity was 6/9 in both eyes oculus uterque (OU), and intraocular pressure was 12 mmHg OU. Eyelids of both eyes revealed round or oval bead-like excrescences that were yellow-white in color, resembling string of pearls involving upper and lower eyelids, suggestive of moniliform blepharosis that is pathognomonic [Figures 1-3].
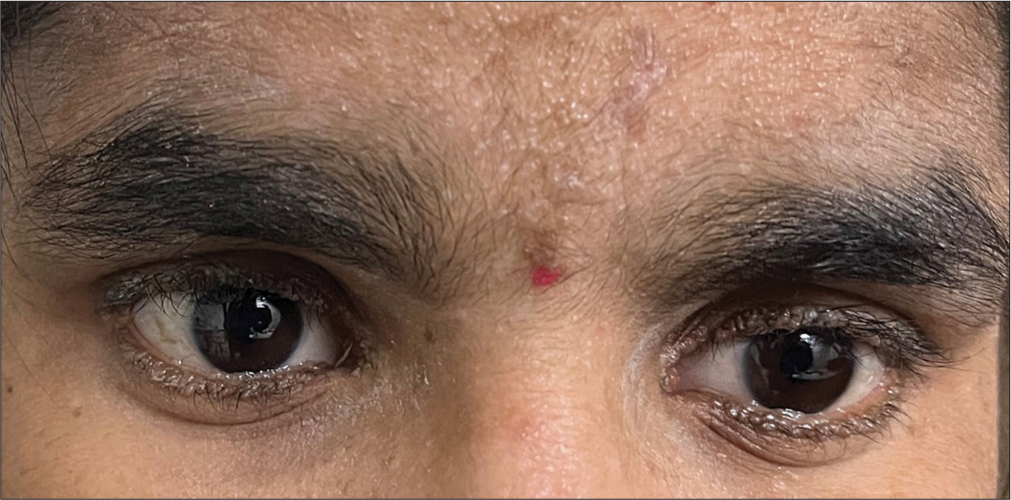
- Moniliform blepharosis.

- Lesions resembling string of pearls.

- Slit-lamp view of moniliform blepharosis.
Anterior segment and fundus evaluation of both eyes were within normal limits. Her systemic examination revealed scars over the dorsum of hands. She had a previous history of warty papule-like lesions on the dorsum of hands and axilla for which she had sought treatment from a dermatologist. She also presented with hoarseness of voice and speech difficulties. Her oral examination revealed a thickened tongue with teeth indentations [Figure 4]. Laryngoscopy revealed a thickening of vocal cords. Her routine baseline investigations were normal.
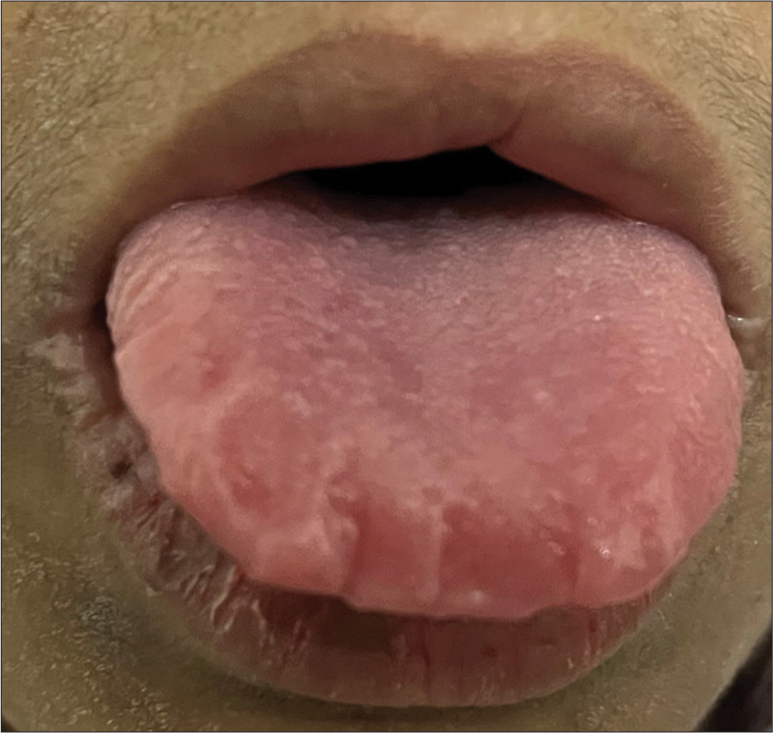
- Thickened tongue.
The patient has been managed conservatively with lid hygiene, artificial tear supplements, topical antibiotics, and antihistamine to relieve itching and prevent the possibility of superimposed bacterial infection. Since there is no permanent cure for this condition, symptomatic treatment remains the mainstay of management.
Her father had been further advised and counseled about the chronic nature of the disease and the need for follow-up with a multidisciplinary team of specialists including an ophthalmologist, otolaryngologist, neurologist, and dentist.
DISCUSSION
LP is a rare, genetic (autosomal recessive) disorder characterized by deposits of hyaline-like material in various organs which include the skin, larynx, oral cavity, as well as internal organs. The skin lesions are seen as blisters in early childhood which get eroded followed by crusting on trivial trauma. Acneiform scarring that is pox-like is particularly evident on the face as well as extremities.
In our case, she had blisters since the age of 8 years for which she had seeked treatment from a dermatologist, eventually seen as healed scars.
The classic pathognomonic sign is the presence of moniliform blepharosis (beaded papules involving the eyelids). The papular infiltration might be subtle in some patients.[4] Oral mucosal infiltration may present as thickening of the lingual frenulum that may cause an inability to protrude the tongue.[4,5] Oral erosions may be noted early in life. In our case, she had hoarseness of voice since childhood and her laryngoscopy revealed thickening of vocal cords. LP is an important clinical entity that is often missed. This case highlights the importance of a detailed ocular evaluation to diagnose a rare systemic genetic disorder.
The treatment of LP consists of a multidisciplinary approach involving an otolaryngologist, dermatologist, ophthalmologist, dentist, and geneticist. The aim of treatment of cutaneous lesions is to provide symptomatic relief and cosmetic care to the patient. The options include acitretin, oral and topical steroids, oral dimethylsulfoxide, and D-penicillamine. The surgical modalities include carbon dioxide laser or cryotherapy depending on the organ system involved. An important role of a geneticist is to provide counseling, especially in the reproductive population.
The prognosis of LP is excellent provided that the disease is diagnosed early and the patient receives specialized care and counseling.
CONCLUSION
LP is an important but frequently ignored/missed entity. It is important that clinicians are aware of this entity so that prompt treatment can be provided to decrease the morbidity of the patient.
Ethical approval
Not applicable.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Urbach-Wiethe disease (lipoglycoproteinosis; lipoid proteinosis; hyalinosis cutis et mucosae). A review. Acta Derm Venereol Suppl (Stockh). 1973;53:1-52.
- [Google Scholar]
- Lipoid proteinosis of the larynx: A cause of voice change in the infant and young child. Int J Pediatr Otorhinolaryngol. 1988;15:33-8.
- [CrossRef] [PubMed] [Google Scholar]
- Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ECM1) Hum Mol Genet. 2002;11:833-40.
- [CrossRef] [PubMed] [Google Scholar]
- Lipoid proteinosis: Clinical, histologic, and ultrastructural investigations. Cutis. 1995;56:220-4.
- [Google Scholar]
- Oral, pharyngeal and laryngeal manifestations in Urbach-Wiethe disease. Ann Clin Res. 1977;9:1-7.
- [Google Scholar]




